class: center, middle, inverse, title-slide .title[ # Taller de análisis de datos de expresión diferencial ] .author[ ### Rodolfo L. Chavez Dominguez ] .date[ ### 3 May 2022 ] --- <style type="text/css"> /* From https://github.com/yihui/xaringan/issues/147 */ .scroll-output { height: 80%; overflow-y: scroll; } /* https://stackoverflow.com/questions/50919104/horizontally-scrollable-output-on-xaringan-slides */ pre { max-width: 100%; overflow-x: scroll; } .remark-slide-content { font-size: 28px; padding: 20px 80px 20px 80px; } .remark-code, .remark-inline-code { background: #f0f0f0; } .remark-code { font-size: 24px; } .huge .remark-code { /*Change made here*/ font-size: 200% !important; } .tiny .remark-code { /*Change made here*/ font-size: 80% !important; } .center2 { margin: 0; position: absolute; top: 50%; left: 50%; -ms-transform: translate(-50%, -50%); transform: translate(-50%, -50%); } /* From https://github.com/garthtarr/sydney_xaringan */ blockquote, .blockquote { display: block; margin-top: 0.1em; margin-bottom: 0.2em; margin-left: 5px; margin-right: 5px; border-left: solid 10px #0148A4; border-top: solid 2px #0148A4; border-bottom: solid 2px #0148A4; border-right: solid 2px #0148A4; box-shadow: 0 0 6px rgba(0,0,0,0.5); /* background-color: #e64626; */ color: #e64626; padding: 0.5em; -moz-border-radius: 5px; -webkit-border-radius: 5px; } /* From https://github.com/garthtarr/sydney_xaringan */ .content-box-blue, .content-box-gray, .content-box-grey, .content-box-army, .content-box-green, .content-box-purple, .content-box-red, .content-box-yellow { box-sizing: border-box; border-radius: 15px; margin: 0 0 15px; overflow: hidden; padding: 0px 20px 0px 20px; width: 100%; } </style> ## Material Este material está basado en el pipeline de [Michael Love](https://www.bioconductor.org/packages/devel/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html) de Bioconductor. <img src="https://images.squarespace-cdn.com/content/v1/5423875be4b03f0c482a58c4/1532953317705-W6TYTE70KG5E6KQEZU1K/ke17ZwdGBToddI8pDm48kNVP8RwsgCc7XlHc2zPQeqdZw-zPPgdn4jUwVcJE1ZvWQUxwkmyExglNqGp0IvTJZUJFbgE-7XRK3dMEBRBhUpxA5wn4368HhrDKfvXqORu9oTEmTJkjlQ2gdQcVngofpWQcE5w-MKnvigIPwIMqpXs/Bioconductor.png" width="300px" style="display: block; margin: auto;" /> --- class: inverse, center, middle # 1: Introducción al RNA-seq --- ## Transcriptómica Estudio de la expresión genética - ¿Cuánto RNA hay? <img src="./Images/transcriptomica.png" width="900px" style="display: block; margin: auto;" /> --- ## RNA-seq <img src="./Images/RNA_isolation.jpeg" width="500px" class="center"> </img> --- ## Aplicaciones <img src="./Images/book.png" width="570px" style="display: block; margin: auto;" /> --- ## Bases de datos <img src="./Images/book.png" width="450px" style="display: block; margin: auto;" /> --- class: inverse, center, middle # 2: Experimento --- ## Experimento Número de acceso GEO: GSE136011 <img src="./Images/NRF2.png" width="800px" style="display: block; margin: auto;" /> --- class: inverse, center, middle # 3: Flujo de trabajo para el análisis de RNA-seq --- ## Flujo general de trabajo <img src="./Images/RNAseq_workflow.png" width="550px" style="display: block; margin: auto;" /> --- ## Control de calidad <img src="./Images/step1.png" width="540px" style="display: block; margin: auto;" /> --- ## Alineamiento y cuantificación <img src="./Images/step2.png" width="950px" style="display: block; margin: auto;" /> --- ## Expresión diferencial y análisis funcionales <img src="./Images/step3.png" width="750px" style="display: block; margin: auto;" /> --- ## Flujo general de trabajo <img src="./Images/RNAseq_workflow.png" width="550px" style="display: block; margin: auto;" /> --- class: inverse, center, middle # 4: Alineamiento --- <img src="./Images/RNAseq_workflow2.png" width="600px" style="display: block; margin: auto;" /> --- ## Alineamiento <img src="./Images/ReadAlignment.png" width="600px" style="display: block; margin: auto;" /> Buscar la región del genoma/transcriptoma a partir de la cual se originaron las lecturas --- ## Alineamiento ## Spliced reads <img src="./Images/SplicedReads2.png" width="600px" style="display: block; margin: auto;" /> --- ## Splice aware - TopHat 2 <img src="./Images/SpliceAware.png" width="600px" style="display: block; margin: auto;" /> -- - HISAT <img src="./Images/HISAT.png" width="600px" style="display: block; margin: auto;" /> -- - STAR <img src="./Images/STAR.png" width="400px" style="display: block; margin: auto;" /> --- ## Alineamiento ## Pseudo-alineadores Pseudo-alineadores (quasi-alineadores): - Kallisto - Sailfish - **Salmon** <!-- --> --- ## Alineamiento ## Pseudo-alineadores <img src="./Images/Pseudoalignment.png" width="600px" style="display: block; margin: auto;" /> --- ## Alineamiento ## Pseudo-alineadores <img src="./Images/PseudoAlignment1.png" width="500px" style="display: block; margin: auto;" /> -- <img src="./Images/PseudoAlignment2.png" width="500px" style="display: block; margin: auto;" /> --- ## Alineamiento ## Pseudo-alineadores <img src="./Images/PseudoAlignment3.png" width="600px" style="display: block; margin: auto;" /> --- class: inverse, center, middle # 5: Tximeta y Tximport --- ## Recordatorio ¿Dónde estamos? <img src="./Images/RNAseq_workflow2.png" width="450px" style="display: block; margin: auto;" /> --- ## Recordatorio ¿Dónde estamos? <img src="Images/worflowTximeta.png" width="450px" style="display: block; margin: auto;" /> --- ## Tximeta Paquetería que conserva <img src="https://journals.plos.org/ploscompbiol/article/figure/image?size=large&id=10.1371/journal.pcbi.1007664.g001" width="650px" style="display: block; margin: auto;" /> --- ## Importar datos Utilizamos `tximeta` para importar los datos. Para ello requerímos de una tabla de metadatos con información de las muestras ```bash #La tabla se ubica en el directorio de salmon_quants con el nombre de metadata.txt ls ../data/salmon_quants ``` ``` ## metadata.txt ## samp10_quant ## samp11_quant ## samp12_quant ## samp1_quant ## samp2_quant ## samp3_quant ## samp4_quant ## samp5_quant ## samp6_quant ## samp7_quant ## samp8_quant ## samp9_quant ``` --- class: center, middle background-image: url("./Images/hands_on.png") background-size: 90% 80% --- ## Importar datos .scroll-output[ Cargamos los datos utilizando el objeto de R .red[*SummarizedExperiment.R*] ```r load("../data/SummarizedExperimentObj.rdata") ``` ] --- class: inverse, center, middle # 6: Objeto Summarized Experiment --- .center2[ <img src="./Images/Slots.png" width="700px" /> ] --- ## Summarized Experiment <img src="./Images/Summarized_experiment.png" width="400px" style="display: block; margin: auto;" /> --- ## Summarized Experiment <img src="./Images/ColData.png" width="400px" style="display: block; margin: auto;" /> --- ## Summarized Experiment ## ColData .scroll-output[ - El cajón o *slot* correspondiente a `ColData` contiene la tabla de metadatos `coldata` empleada para importar las cuentas con `tximeta` - Para acceder a **.red[ColData]** usar el siguiente comando: ```r colData(se) ``` ``` ## DataFrame with 12 rows and 8 columns ## file_name ## <factor> ## siRNA_NRF2_1 SRR10004274_GSM4039376_NRF2_siRNA_rep_1_Homo_sapiens_RNA-Seq_1.fastq.gz ## siRNA_control_1 SRR10004275_GSM4039377_NRF2_siRNA_cont_rep1_Homo_sapiens_RNA-Seq_1.fastq.gz ## siRNA_NRF2_2 SRR10004276_GSM4039378_NRF2_siRNA_rep_2_Homo_sapiens_RNA-Seq_1.fastq.gz ## siRNA_control_2 SRR10004277_GSM4039379_NRF2_siRNA_cont_rep2_Homo_sapiens_RNA-Seq_1.fastq.gz ## siRNA_NRF2_3 SRR10004278_GSM4039380_NRF2_siRNA_rep_3_Homo_sapiens_RNA-Seq_1.fastq.gz ## ... ... ## mut_NRF2_1 SRR10004281_GSM4039383_NRF2_mutant_rep_1_Homo_sapiens_RNA-Seq_1.fastq.gz ## CT_2 SRR10004282_GSM4039384_NRF2_wild_type_rep_2_Homo_sapiens_RNA-Seq_1.fastq.gz ## mut_NRF2_2 SRR10004283_GSM4039385_NRF2_mutant_rep_2_Homo_sapiens_RNA-Seq_1.fastq.gz ## CT_3 SRR10004284_GSM4039386_NRF2_wild_type_rep_3_Homo_sapiens_RNA-Seq_1.fastq.gz ## mut_NRF2_3 SRR10004285_GSM4039387_NRF2_mutant_rep_3_Homo_sapiens_RNA-Seq_1.fastq.gz ## Treatment Replicate unique_id key Geno ## <factor> <integer> <factor> <factor> <factor> ## siRNA_NRF2_1 siRNA_NRF2 1 siRNA_NRF2_1 samp1 WT ## siRNA_control_1 siRNA_control 1 siRNA_control_1 samp2 WT ## siRNA_NRF2_2 siRNA_NRF2 2 siRNA_NRF2_2 samp3 WT ## siRNA_control_2 siRNA_control 2 siRNA_control_2 samp4 WT ## siRNA_NRF2_3 siRNA_NRF2 3 siRNA_NRF2_3 samp5 WT ## ... ... ... ... ... ... ## mut_NRF2_1 mut_NRF2 1 mut_NRF2_1 samp8 Mut ## CT_2 CT 2 CT_2 samp9 WT ## mut_NRF2_2 mut_NRF2 2 mut_NRF2_2 samp10 Mut ## CT_3 CT 3 CT_3 samp11 WT ## mut_NRF2_3 mut_NRF2 3 mut_NRF2_3 samp12 Mut ## exist names ## <logical> <character> ## siRNA_NRF2_1 TRUE samp1_quant ## siRNA_control_1 TRUE samp2_quant ## siRNA_NRF2_2 TRUE samp3_quant ## siRNA_control_2 TRUE samp4_quant ## siRNA_NRF2_3 TRUE samp5_quant ## ... ... ... ## mut_NRF2_1 TRUE samp8_quant ## CT_2 TRUE samp9_quant ## mut_NRF2_2 TRUE samp10_quant ## CT_3 TRUE samp11_quant ## mut_NRF2_3 TRUE samp12_quant ``` ] --- ## Summarized Experiment ## ColData .scroll-output[ - El slot **ColData** es un objeto con clase de *DataFrame* - *Rownames* de **ColData** corresponden a los *Colnames* en el slot **Assay**- **.red[Importante para análisis con DESeq2]** ] --- ## Summarized experiment <img src="./Images/RowRanges.png" width="400px" style="display: block; margin: auto;" /> --- ## Summarized experiment ## rowRanges .scroll-output[ - El cajón de `rowRanges` es un objeto de tipo *GRanges* que hace referencia a las coordenadas de cada transcrito y su anotación correspondiente - Para acceder al **.red[rowRanges]** usar: ```r rowRanges(se) ``` ``` ## GRanges object with 184844 ranges and 9 metadata columns: ## seqnames ranges strand | ## <Rle> <IRanges> <Rle> | ## ENST00000631435 CHR_HSCHR7_2_CTG6 142847306-142847317 + | ## ENST00000415118 14 22438547-22438554 + | ## ENST00000434970 14 22439007-22439015 + | ## ENST00000448914 14 22449113-22449125 + | ## ENST00000632524 CHR_HSCHR14_3_CTG1 105866322-105866332 - | ## ... ... ... ... . ## ENST00000444456 1 247974741-247975653 + | ## ENST00000416377 1 248003129-248004068 + | ## ENST00000318104 1 248121932-248122882 + | ## ENST00000606902 1 248498308-248498649 + | ## ENST00000427827 1 248549444-248549762 - | ## tx_id tx_biotype tx_cds_seq_start ## <character> <character> <integer> ## ENST00000631435 ENST00000631435 TR_D_gene 142847306 ## ENST00000415118 ENST00000415118 TR_D_gene 22438547 ## ENST00000434970 ENST00000434970 TR_D_gene 22439007 ## ENST00000448914 ENST00000448914 TR_D_gene 22449113 ## ENST00000632524 ENST00000632524 IG_D_gene 105866322 ## ... ... ... ... ## ENST00000444456 ENST00000444456 unprocessed_pseudogene <NA> ## ENST00000416377 ENST00000416377 unprocessed_pseudogene <NA> ## ENST00000318104 ENST00000318104 unprocessed_pseudogene <NA> ## ENST00000606902 ENST00000606902 unprocessed_pseudogene <NA> ## ENST00000427827 ENST00000427827 unprocessed_pseudogene <NA> ## tx_cds_seq_end gene_id tx_support_level ## <integer> <character> <integer> ## ENST00000631435 142847317 ENSG00000282253 <NA> ## ENST00000415118 22438554 ENSG00000223997 <NA> ## ENST00000434970 22439015 ENSG00000237235 <NA> ## ENST00000448914 22449125 ENSG00000228985 <NA> ## ENST00000632524 105866332 ENSG00000282455 <NA> ## ... ... ... ... ## ENST00000444456 <NA> ENSG00000237492 <NA> ## ENST00000416377 <NA> ENSG00000232215 <NA> ## ENST00000318104 <NA> ENSG00000177233 <NA> ## ENST00000606902 <NA> ENSG00000271934 <NA> ## ENST00000427827 <NA> ENSG00000227102 <NA> ## tx_id_version gc_content tx_name ## <character> <numeric> <character> ## ENST00000631435 ENST00000631435.1 83.3333 ENST00000631435 ## ENST00000415118 ENST00000415118.1 25.0000 ENST00000415118 ## ENST00000434970 ENST00000434970.2 55.5556 ENST00000434970 ## ENST00000448914 ENST00000448914.1 61.5385 ENST00000448914 ## ENST00000632524 ENST00000632524.1 54.5455 ENST00000632524 ## ... ... ... ... ## ENST00000444456 ENST00000444456.1 44.0307 ENST00000444456 ## ENST00000416377 ENST00000416377.2 45.3191 ENST00000416377 ## ENST00000318104 ENST00000318104.2 47.7392 ENST00000318104 ## ENST00000606902 ENST00000606902.2 45.9064 ENST00000606902 ## ENST00000427827 ENST00000427827.2 45.4545 ENST00000427827 ## ------- ## seqinfo: 455 sequences from GRCh38 genome ``` ] --- ## Summarized experiment <img src="./Images/Assay.png" width="400px" style="display: block; margin: auto;" /> --- ## Summarized experiment ## Assay .scroll-output[ - El *slot* `assay` almacena la información de las cuentas para cada transcrito dividida en tres niveles: ```r assayNames(se) ``` ``` ## [1] "counts" "abundance" "length" ``` - Para acceder a la matriz de cuentas estimadas por *Salmon*, correr: ```r head(assay(se), 5) ``` ``` ## siRNA_NRF2_1 siRNA_control_1 siRNA_NRF2_2 siRNA_control_2 ## ENST00000631435 0 0 0 0 ## ENST00000415118 0 0 0 0 ## ENST00000434970 0 0 0 0 ## ENST00000448914 0 0 0 0 ## ENST00000632524 0 0 0 0 ## siRNA_NRF2_3 siRNA_control_3 CT_1 mut_NRF2_1 CT_2 mut_NRF2_2 ## ENST00000631435 0 0 0 0 0 0 ## ENST00000415118 0 0 0 0 0 0 ## ENST00000434970 0 0 0 0 0 0 ## ENST00000448914 0 0 0 0 0 0 ## ENST00000632524 0 0 0 0 0 0 ## CT_3 mut_NRF2_3 ## ENST00000631435 0 0 ## ENST00000415118 0 0 ## ENST00000434970 0 0 ## ENST00000448914 0 0 ## ENST00000632524 0 0 ``` ] --- ## Summarized experiment ## Assay - Las matriz de abundancia *(TPM)* puede obtenerse: .code70[ ```r ## Obtener matriz de TPM head(se@assays@data$abundance, 5) ``` ``` ## samp1_quant samp2_quant samp3_quant samp4_quant samp5_quant ## ENST00000631435 0 0 0 0 0 ## ENST00000415118 0 0 0 0 0 ## ENST00000434970 0 0 0 0 0 ## ENST00000448914 0 0 0 0 0 ## ENST00000632524 0 0 0 0 0 ## samp6_quant samp7_quant samp8_quant samp9_quant samp10_quant ## ENST00000631435 0 0 0 0 0 ## ENST00000415118 0 0 0 0 0 ## ENST00000434970 0 0 0 0 0 ## ENST00000448914 0 0 0 0 0 ## ENST00000632524 0 0 0 0 0 ## samp11_quant samp12_quant ## ENST00000631435 0 0 ## ENST00000415118 0 0 ## ENST00000434970 0 0 ## ENST00000448914 0 0 ## ENST00000632524 0 0 ``` ] --- ## Summarized experiment .scroll-output[ ¿Recuerdan a qué tiene que ser igual *Rownames* del slot colData? .code90[ ```r rownames(colData(se)) ``` ``` ## [1] "siRNA_NRF2_1" "siRNA_control_1" "siRNA_NRF2_2" "siRNA_control_2" ## [5] "siRNA_NRF2_3" "siRNA_control_3" "CT_1" "mut_NRF2_1" ## [9] "CT_2" "mut_NRF2_2" "CT_3" "mut_NRF2_3" ``` ```r colnames(assay(se)) ``` ``` ## [1] "siRNA_NRF2_1" "siRNA_control_1" "siRNA_NRF2_2" "siRNA_control_2" ## [5] "siRNA_NRF2_3" "siRNA_control_3" "CT_1" "mut_NRF2_1" ## [9] "CT_2" "mut_NRF2_2" "CT_3" "mut_NRF2_3" ``` ```r ## Comprobar que rownames de colData es igual a colnames de assay row.names(colData(se)) == colnames(assay(se)) ``` ``` ## [1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE ``` ```r ##Para obtener las cuentas a nivel de gen utiliza gse <- summarizeToGene(se) ``` ] ] --- class: inverse, center, middle # 7: Exploración de los datos --- .center2[ <img src="./Images/PCAplot.png" width="700px" /> ] --- ## Exploración de datos ## ¿Por qué es importante explorar los datos? -- - Paso previo al análisis de expresión diferencial -- - Análisis de calidad de los datos -- - Permite conocer la congurencian entre individuos o réplicas -- - Mediante gráficos, visualizar comportamiento de los datos <svg viewBox="0 0 320 512" style="height:1em;position:relative;display:inline-block;top:.1em;" xmlns="http://www.w3.org/2000/svg"> <path d="M285.476 272.971L91.132 467.314c-9.373 9.373-24.569 9.373-33.941 0l-22.667-22.667c-9.357-9.357-9.375-24.522-.04-33.901L188.505 256 34.484 101.255c-9.335-9.379-9.317-24.544.04-33.901l22.667-22.667c9.373-9.373 24.569-9.373 33.941 0L285.475 239.03c9.373 9.372 9.373 24.568.001 33.941z"></path></svg> Outliers --- ## Exploración de datos .pull-left[ <img src="./Images/PCAplot2.png" width="400px" /> ] -- .pull-right[ <img src="./Images/Heatmap.png" width="400px" /> ] --- ## Exploración de datos .pull-left[ <img src="./Images/PCAplot2.png" width="400px" /> **.red[PCA]** ] .pull-right[ <img src="./Images/Heatmap.png" width="400px" /> **.red[Heatmap]** ] --- ## PCA -- - Análisis de componentes principales -- - Método algebraico para reducir la dimensionalidad de juegos de datos complejos (múltiples variables) -- - Reducción de dimensionalidad o variables permite un análisis exploratorio más intuitivo -- .blockquote[Reducción de variables implica preservar y captar la mayor información sobre los datos ] --- ## PCA ## Normalización de los datos -- <img src="./Images/zscore.png" width="350px" style="display: block; margin: auto;" /> Escalación de los datos <svg viewBox="0 0 320 512" style="height:1em;position:relative;display:inline-block;top:.1em;" xmlns="http://www.w3.org/2000/svg"> <path d="M285.476 272.971L91.132 467.314c-9.373 9.373-24.569 9.373-33.941 0l-22.667-22.667c-9.357-9.357-9.375-24.522-.04-33.901L188.505 256 34.484 101.255c-9.335-9.379-9.317-24.544.04-33.901l22.667-22.667c9.373-9.373 24.569-9.373 33.941 0L285.475 239.03c9.373 9.372 9.373 24.568.001 33.941z"></path></svg> Normalización para hacerlos más comparables -- <img src="./Images/covariancematrix.png" width="350px" style="display: block; margin: auto;" /> Busqueda de correlación entre las variables <svg viewBox="0 0 320 512" style="height:1em;position:relative;display:inline-block;top:.1em;" xmlns="http://www.w3.org/2000/svg"> <path d="M285.476 272.971L91.132 467.314c-9.373 9.373-24.569 9.373-33.941 0l-22.667-22.667c-9.357-9.357-9.375-24.522-.04-33.901L188.505 256 34.484 101.255c-9.335-9.379-9.317-24.544.04-33.901l22.667-22.667c9.373-9.373 24.569-9.373 33.941 0L285.475 239.03c9.373 9.372 9.373 24.568.001 33.941z"></path></svg> correlación positiva o negativa --- ## PCA .center[  ] -- .blockquote[Los componentes principales son los nuevos ejes que maximizan la distancia de los datos al origen ] --- ## PCA ## Componentes principales -- - El .green[primer componente] es aquel en el que los datos presentan la mayor separación (variación) -- - El .orange[segundo componente] es el que tiene la segunda mayor separación entre los datos y es perpendicular al primer componente -- - ¿Cuantos componentes existen? <svg viewBox="0 0 320 512" style="height:1em;position:relative;display:inline-block;top:.1em;" xmlns="http://www.w3.org/2000/svg"> <path d="M285.476 272.971L91.132 467.314c-9.373 9.373-24.569 9.373-33.941 0l-22.667-22.667c-9.357-9.357-9.375-24.522-.04-33.901L188.505 256 34.484 101.255c-9.335-9.379-9.317-24.544.04-33.901l22.667-22.667c9.373-9.373 24.569-9.373 33.941 0L285.475 239.03c9.373 9.372 9.373 24.568.001 33.941z"></path></svg> tantas variables en el juego de datos --- ## PCA ## Componentes principales - Cada componente principal tiene asociado un eigenvector (vector unitario) y un eigenvalue (cantidad escalar) -- - Los eigenvalues son la suma del cuadrado de las distancias de los puntos proyectados sobre dicho componente principal -- - Los eigenvalues permiten seleccionar cuál es el componente principal que explica la mayor variación en los datos --- ## PCA <img src="./Images/Screeplot.png" width="450px" style="display: block; margin: auto;" /> Screeplot --- ## PCA - En sets de datos de RNAseq, el PCA permite visualizar la distancia o congruencia de los datos -- - Generalmente son gráficas de puntos (muestras) en dos dimensiones (dos componentes) que resumen las principales fuentes de varianza -- .center[ <img src="./Images/PCARNAseq.png" width="400px" /> ] --- class: center, middle background-image: url("./Images/hands_on.png") background-size: 90% 80% --- ## Exploración de los datos ### Creación del objeto DDS .scroll-output[ Crear un objeto `DESeq` (*SummarizedExperiment*) a partir del objeto `gse` ```r dds <- DESeqDataSet(se = gse, ##Objeto SummarizedExperiment que contiene la matriz de cuentas crudas design = ~ Treatment) ##Variable con los grupos experimentales a contrastar. Forma parte de las columnas de coldata ``` ``` ## using counts and average transcript lengths from tximeta ``` Visualizar el grupo de referencia o **control** de la variable seleccionada ```r ##El primer nivel o elemento es el grupo control ("CT") levels(dds$Treatment) ``` ``` ## [1] "CT" "mut_NRF2" "siRNA_control" "siRNA_NRF2" ``` Dependiendo de la pregunta a responder, recodifica el grupo de referencia: ```r dds$Treatment <- relevel(dds$Treatment, "siRNA_control") levels(dds$Treatment) ``` ``` ## [1] "siRNA_control" "CT" "mut_NRF2" "siRNA_NRF2" ``` Selecciona aquellos genes que muestran mayor abundancia usando un punto de corte ```r ##Recupera los genes que muestran más de 1 cuenta en al menos 3 muestras keep <- rowSums(counts(dds) >= 1) >=3 ##Visualiza cuántos genes pasaron el filtro de abundancia table(keep) ``` ``` ## keep ## FALSE TRUE ## 16846 21299 ``` ```r ##Corta los genes que pasaron el filtro de abundancia del objeto dds dds <- dds[keep, ] ``` ] --- ## Exploración de los datos ### PCA .scroll-output[ 5. Para explorar los datos por medio del PCA necesitamos normalizar las cuentas crudas para que los datos sean más comparables entre sí. Empleamos la transformación *regularized-logarithm transformation* recomendada para juegos de datos con una n < 30: ```r rld <- rlog(dds, blind = F) ``` ``` ## using 'avgTxLength' from assays(dds), correcting for library size ``` 6. Realiza el PCA empleando el paquete `PCAtools`: ```r ##Indicarle a la función pca que los datos deben de ser escalados para que sean más comparables entre sí pca <- pca(assay(rld), metadata = colData(rld), scale = T) ##Graficar el resultado del PCA mostrando las etiquetas de acuerdo al nombr + tratamiento + réplica y coloreando los puntos de acuerdo al tipo de tratamiento biplot(pca, lab = rownames(colData(rld)), colby = "Treatment") ``` <!-- --> ] --- class: inverse, center, middle # 8: Análisis de expresión diferencial --- ## Análisis de expresión diferencial (DE) Prueba estadística para obtener los genes que varían entre las condiciones experimentales <img src="https://raw.githubusercontent.com/hbctraining/DGE_workshop/master/img/de_theory.png" width="750px" style="display: block; margin: auto;" /> --- ## Pasos en el DE <img src="https://github.com/hbctraining/DGE_workshop/raw/master/img/deseq2_workflow_separate.png" width="350px" style="display: block; margin: auto;" /> --- ## Normalización Es el primer paso del análisis de expresión diferencial y es necesario para realizar comparaciones acertadas entre muestras. -- Las cuentas crudas están conformadas por un componente **"interesante"** (la expresión de RNA) y componentes "no interesantes" (como los efectos de lote, ruido de la plataforma, etc.). -- La normalzación escala las cuentas para tratar de reducir los componentes "no interesantes" y poder comparar las muestras entre si. --- ## Criteríos para normalizar Se puede normalizar considerando: - La profundidad (tamaño de librería) <img src="https://hbctraining.github.io/DGE_workshop/img/normalization_methods_depth.png" width="450px" style="display: block; margin: auto;" /> --- ## Criteríos para normalizar Se puede normalizar considerando: - El tamaño del gen <img src="https://hbctraining.github.io/DGE_workshop/img/normalization_methods_length.png" width="450px" style="display: block; margin: auto;" /> --- ## Criteríos para normalizar Se puede normalizar considerando: - Composición de RNA <img src="https://hbctraining.github.io/DGE_workshop/img/normalization_methods_composition.png" width="350px" style="display: block; margin: auto;" /> --- ## Métodos comunes .scroll-output[ | Método de normalización | Descripción | Factores de evaluación | Recomendaciones de uso | |:-----------------------------------------------------------------------------------:|:----------------------------------------------------------------------------------------------------------------------------:|:--------------------------------------------------:|:-------------------------------------------------------------------------------------------------------------------:| | CPM (cuentas por millón): cuentas escalados por el número total de lecturas | profundidad de secuenciación | comparaciones de cuentas de genes entre réplicas del mismo grupo de muestras| NO para comparaciones dentro de la muestra o análisis de DE.| | TPM (transcritos por kilobase por millón de lecturas): cuentas por longitud de transcripción (kb) por millón de lecturas mapeadas | profundidad de secuenciación y longitud de genes| comparaciones de cuentas de genes dentro de una muestra o entre muestras del mismo grupo de muestras| NO para análisis de DE. | | RPKM/FPKM (lecturas/fragmentos por kilobase de exón por millón de lecturas/fragmentos mapeados| similar a TPM, profundidad de secuenciación y longitud del gen | comparaciones de cuentas entre genes dentro de una muestra | NO para comparaciones entre muestras o análisis de DE.| | Mediana de ratios de DESeq2 | cuentas divididas por factores de tamaño específicos de la muestra determinados por la mediana del ratio de cuentas de genes en relación con la media geométrica por gen | profundidad de secuenciación y composición del RNA | comparaciones de cuentas de genes entre muestras y para el análisis de DE; NO para comparaciones dentro de la muestra | | La media cortada de los valores M de EdgeR (TMM) | utiliza una media recortada ponderada de los ratios de expresión logarítmica entre las muestras | profundidad de secuenciación | composición de RNA y longitud de los genes. | ] --- ## DESeq2 - Normalización para análisis de expresión diferencial: - Factor de normalización de `DESeq2` - Normalización para visualización u otras aplicaciones: - Variance stabilizing transformation (VST) - Regularized-logarithm transformation (rlog) --- ## Normalización por ratios de medias geométricas (DESeq2) .scroll-output[ `DESeq2`ajusta a un modelo lineal generalizado (GLM) de la familia binomial negativa (NB). ``` ## # A tibble: 2 × 3 ## gene muestraA muestraB ## <chr> <dbl> <dbl> ## 1 gene1 1749 943 ## 2 gene2 35 29 ``` 1. Crea una pseudo-referencia por muestra (promedio geometrico por fila) `sqrt(muestraA * muestra B)` ``` ## # A tibble: 2 × 4 ## # Rowwise: ## gene muestraA muestraB prom_geom ## <chr> <dbl> <dbl> <dbl> ## 1 gene1 1749 943 1284. ## 2 gene2 35 29 31.9 ``` 2. Se calcula la fración `muestra/pseudo-referencia` ``` ## # A tibble: 2 × 6 ## # Rowwise: ## gene muestraA muestraB prom_geom muestraA_pseudo_ref muestraB_pseudo_ref ## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> ## 1 gene1 1749 943 1284. 1.36 0.734 ## 2 gene2 35 29 31.9 1.10 0.910 ``` 3. Se calcula un factor de normalización (size factor) utilizando la `mediana` por columnas. ``` ## [1] 1.230235 ``` ``` ## [1] 0.8222689 ``` 4. Se dividen las `cuentas crudas/size factor` para calcular las cuentas normalizadas. ``` ## # A tibble: 2 × 3 ## # Rowwise: ## gene muestraA muestraB ## <chr> <dbl> <dbl> ## 1 gene1 1749 943 ## 2 gene2 35 29 ``` ``` ## # A tibble: 2 × 2 ## # Rowwise: ## norm_muestraA norm_muestraB ## <dbl> <dbl> ## 1 1422. 1147. ## 2 28.4 35.3 ``` ] --- ## Otras transformaciones .scroll-output[ Puedes realizar otras transformaciones en `DESeq2` para estabilizar la varianza a través de los differentes valores promedio de expresión. <!-- --><!-- --> ```r # variance stabilizing transformation (VST), (Anders and Huber 2010) vsd <- vst(dds, blind = FALSE) ``` ``` ## using 'avgTxLength' from assays(dds), correcting for library size ``` ```r head(assay(vsd), 3) ``` ``` ## siRNA_NRF2_1 siRNA_control_1 siRNA_NRF2_2 siRNA_control_2 ## ENSG00000000003 11.67435 11.63822 11.72695 11.73399 ## ENSG00000000419 11.71082 11.78356 11.74230 11.78642 ## ENSG00000000457 10.52426 10.50532 10.46261 10.51811 ## siRNA_NRF2_3 siRNA_control_3 CT_1 mut_NRF2_1 CT_2 ## ENSG00000000003 11.84279 11.68878 11.14964 11.17933 11.09670 ## ENSG00000000419 11.80449 11.70709 11.48465 11.46978 11.40275 ## ENSG00000000457 10.54239 10.52716 10.46414 10.47452 10.46257 ## mut_NRF2_2 CT_3 mut_NRF2_3 ## ENSG00000000003 11.24472 11.17600 11.12306 ## ENSG00000000419 11.46835 11.50914 11.50147 ## ENSG00000000457 10.50666 10.46139 10.45044 ``` ```r # regularized-logarithm transformation (rlog), (Love, Huber, and Anders 2014) rld <- rlog(dds, blind = FALSE) ``` ``` ## using 'avgTxLength' from assays(dds), correcting for library size ``` ```r head(assay(rld), 3) ``` ``` ## siRNA_NRF2_1 siRNA_control_1 siRNA_NRF2_2 siRNA_control_2 ## ENSG00000000003 10.876558 10.819047 10.958594 10.969500 ## ENSG00000000419 10.944160 11.056010 10.992956 11.060526 ## ENSG00000000457 8.345928 8.289919 8.160224 8.327933 ## siRNA_NRF2_3 siRNA_control_3 CT_1 mut_NRF2_1 CT_2 ## ENSG00000000003 11.134274 10.899100 9.957696 10.016126 9.851707 ## ENSG00000000419 11.087811 10.938303 10.577168 10.551995 10.436115 ## ENSG00000000457 8.398004 8.354244 8.165047 8.197047 8.160009 ## mut_NRF2_2 CT_3 mut_NRF2_3 ## ENSG00000000003 10.140861 10.009398 9.905331 ## ENSG00000000419 10.549557 10.618447 10.605587 ## ENSG00000000457 8.293887 8.156555 8.122530 ``` ```r # Normalización con el factor de normalizacion dds <- estimateSizeFactors(dds) ``` ``` ## using 'avgTxLength' from assays(dds), correcting for library size ``` ```r # Juntar los datos de las tres normalizaciones df <- bind_rows( as_data_frame(log2(counts(dds, normalized=TRUE)[, 1:2]+1)) %>% mutate(transformation = "log2(x + 1)"), as_data_frame(assay(vsd)[, 1:2]) %>% mutate(transformation = "vst"), as_data_frame(assay(rld)[, 1:2]) %>% mutate(transformation = "rlog")) ``` ``` ## Warning: `as_data_frame()` was deprecated in tibble 2.0.0. ## Please use `as_tibble()` instead. ## The signature and semantics have changed, see `?as_tibble`. ## This warning is displayed once every 8 hours. ## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was generated. ``` ```r # Renombrar columnas colnames(df)[1:2] <- c("x", "y") # Nombre de las graficas lvls <- c("log2(x + 1)", "vst", "rlog") # Agrupar los tres tipos de normalizacion en grupos como factores df$transformation <- factor(df$transformation, levels=lvls) # Plotear los datos ggplot(df, aes(x = x, y = y)) + geom_hex(bins = 80) + coord_fixed() + facet_grid( . ~ transformation) ``` <!-- --> ] --- ## Dispersión La dispersión es una medida de la variabilidad de los datos como (varianza, sd, etc.). En `DESeq2` se utiliza `α`: $$ \alpha \propto 1/mean $$ $$ \alpha \propto variance $$ Así que **La dispersión es mayor para genes con poca abundancia y menor para genes abundantes**. Y la **dispersión refleja la varianza**. .blockquote[ `DESeq2` asume que los genes con similar expresión tienen similar dispersión ] --- ## Ajustando la dispersión <img src="https://github.com/hbctraining/DGE_workshop/raw/master/img/deseq_dispersion1.png" width="450px" style="display: block; margin: auto;" /> --- ## Reduciendo la dispersión <img src="https://github.com/hbctraining/DGE_workshop/raw/master/img/deseq_dispersion2.png" width="750px" style="display: block; margin: auto;" /> --- class: center, middle background-image: url("./Images/hands_on.png") background-size: 90% 80% --- ## Ejercicio .scroll-output[ La librería de `DESeq2` normaliza y realiza el análisis de expresión diferencial en una sola función. Así que ya realizamos el análisis, vamos a verlo: 1. Corre la función `DESeq()` para normalizar, estimar la dispersión y ajustar los datos al modelo binomial negativo. ```r dds <- DESeq(dds) ``` ``` ## using pre-existing normalization factors ``` ``` ## estimating dispersions ``` ``` ## gene-wise dispersion estimates ``` ``` ## mean-dispersion relationship ``` ``` ## final dispersion estimates ``` ``` ## fitting model and testing ``` 2. Para obtener los resultados del análisis de expresión diferencial usa la función `results` ```r ##Usemos la función results con los parámetros por default res <- results(dds) ##Visualiza el objeto que contiene los resultados res ``` ``` ## log2 fold change (MLE): Treatment siRNA NRF2 vs siRNA control ## Wald test p-value: Treatment siRNA NRF2 vs siRNA control ## DataFrame with 21299 rows and 6 columns ## baseMean log2FoldChange lfcSE stat pvalue ## <numeric> <numeric> <numeric> <numeric> <numeric> ## ENSG00000000003 1497.757504 0.0982849 0.0902643 1.088857 2.76217e-01 ## ENSG00000000419 1789.280526 -0.0107935 0.0702472 -0.153650 8.77885e-01 ## ENSG00000000457 304.687311 -0.0243559 0.1151762 -0.211466 8.32523e-01 ## ENSG00000000460 467.602306 0.5142010 0.1159843 4.433368 9.27721e-06 ## ENSG00000000938 0.618937 1.3390032 2.8076874 0.476906 6.33429e-01 ## ... ... ... ... ... ... ## ENSG00000288690 8.913454 0.0326943 0.663455 0.0492789 0.960697 ## ENSG00000288694 0.258397 0.8179394 4.365460 0.1873661 0.851374 ## ENSG00000288695 11.159112 -0.3769746 0.701013 -0.5377567 0.590745 ## ENSG00000288698 23.403296 0.5109625 1.060520 0.4818036 0.629946 ## ENSG00000288699 0.869679 1.5093784 3.007207 0.5019203 0.615724 ## padj ## <numeric> ## ENSG00000000003 0.476825323 ## ENSG00000000419 0.948127083 ## ENSG00000000457 0.924197635 ## ENSG00000000460 0.000121719 ## ENSG00000000938 NA ## ... ... ## ENSG00000288690 0.980632 ## ENSG00000288694 NA ## ENSG00000288695 0.768387 ## ENSG00000288698 0.795601 ## ENSG00000288699 NA ``` ```r ##Obten el número de genes sobre- y sub-expresados de acuerdo a la prueba estadística summary(res) ``` ``` ## ## out of 21299 with nonzero total read count ## adjusted p-value < 0.1 ## LFC > 0 (up) : 2271, 11% ## LFC < 0 (down) : 2660, 12% ## outliers [1] : 26, 0.12% ## low counts [2] : 5778, 27% ## (mean count < 8) ## [1] see 'cooksCutoff' argument of ?results ## [2] see 'independentFiltering' argument of ?results ``` 3. Explora el objeto para tener información del contenido de cada columna: ```r mcols(res, use.names = TRUE) ``` ``` ## DataFrame with 6 rows and 2 columns ## type description ## <character> <character> ## baseMean intermediate mean of normalized c.. ## log2FoldChange results log2 fold change (ML.. ## lfcSE results standard error: Trea.. ## stat results Wald statistic: Trea.. ## pvalue results Wald test p-value: T.. ## padj results BH adjusted p-values ``` ] --- ## Análisis de expresión diferencial **_Warning_** .content-box-red[Los parámetros por default de la función `res()` hacen referencia a la siguiente hipótesis nula:] .blockquote[La expresión del Gen X en la condición problema es igual a la condición control] Si deseamos modificar el *cutoff* o valor de corte del log2FoldChange: --- .scroll-output[ ```r ##Emplea los argumentos lfcthreshold y alpha para especificar ambos puntos de corte de forma simultánea res_LFC <- results(dds, lfcThreshold = 1, alpha = 0.05) res_LFC ``` ``` ## log2 fold change (MLE): Treatment siRNA NRF2 vs siRNA control ## Wald test p-value: Treatment siRNA NRF2 vs siRNA control ## DataFrame with 21299 rows and 6 columns ## baseMean log2FoldChange lfcSE stat pvalue ## <numeric> <numeric> <numeric> <numeric> <numeric> ## ENSG00000000003 1497.757504 0.0982849 0.0902643 0.000000 1.000000 ## ENSG00000000419 1789.280526 -0.0107935 0.0702472 0.000000 1.000000 ## ENSG00000000457 304.687311 -0.0243559 0.1151762 0.000000 1.000000 ## ENSG00000000460 467.602306 0.5142010 0.1159843 0.000000 1.000000 ## ENSG00000000938 0.618937 1.3390032 2.8076874 0.120741 0.903896 ## ... ... ... ... ... ... ## ENSG00000288690 8.913454 0.0326943 0.663455 0.000000 1.000000 ## ENSG00000288694 0.258397 0.8179394 4.365460 0.000000 1.000000 ## ENSG00000288695 11.159112 -0.3769746 0.701013 0.000000 1.000000 ## ENSG00000288698 23.403296 0.5109625 1.060520 0.000000 1.000000 ## ENSG00000288699 0.869679 1.5093784 3.007207 0.169386 0.865493 ## padj ## <numeric> ## ENSG00000000003 1 ## ENSG00000000419 1 ## ENSG00000000457 1 ## ENSG00000000460 1 ## ENSG00000000938 1 ## ... ... ## ENSG00000288690 1 ## ENSG00000288694 1 ## ENSG00000288695 1 ## ENSG00000288698 1 ## ENSG00000288699 1 ``` ```r summary(res_LFC) ``` ``` ## ## out of 21299 with nonzero total read count ## adjusted p-value < 0.05 ## LFC > 1.00 (up) : 7, 0.033% ## LFC < -1.00 (down) : 8, 0.038% ## outliers [1] : 26, 0.12% ## low counts [2] : 0, 0% ## (mean count < 0) ## [1] see 'cooksCutoff' argument of ?results ## [2] see 'independentFiltering' argument of ?results ``` .content-box-red[ <strong>NUNCA</strong> realices la prueba de expresión diferencial con un valor de corte X (log2FoldChange) y posteriormente filtres los genes diferencialmente expresados empleando otro valor de log2FoldChange. ] ] --- class: inverse, center, middle # 9: Visualización de los resultados --- .center[ ### ¿Cuántos tipos de gráficos conoces para visualizar resultados del análisis DE? ] .pull-left[ <img src="./Images/MAplot.png" width="350px" /> ] .pull-right[ <img src="./Images/Volcanoplot.png" width="350px" /> ] --- .center[ ### ¿Cuántos tipos de gráficos conoces para visualizar resultados del análisis DE? ] .pull-left[ <img src="./Images/MAplot.png" width="350px" /> **.green[MAplot]** ] .pull-right[ <img src="./Images/Volcanoplot.png" width="350px" /> **.green[Volcano plot]** ] --- # MAplot .scroll-output[ Gráfico que representa la distribución de los genes o transcritos en las comparaciones de interés **M** eje y de *minus*: .content-box-blue[$$logTx - logCT = logTx/CT$$ ] **A** eje x de *average* -> Promedio de las cuentas normalizadas para cada gen en las condiciones experimentales de la o las variables de interés Para generar el **MAplot** usa: .code90[ ```r ##Es importante que recuerden que la hipótesis nula que se probó fue ##"El lfc del gen n es igual a 0" por lo tanto los genes coloreados son... plotMA(res) ``` 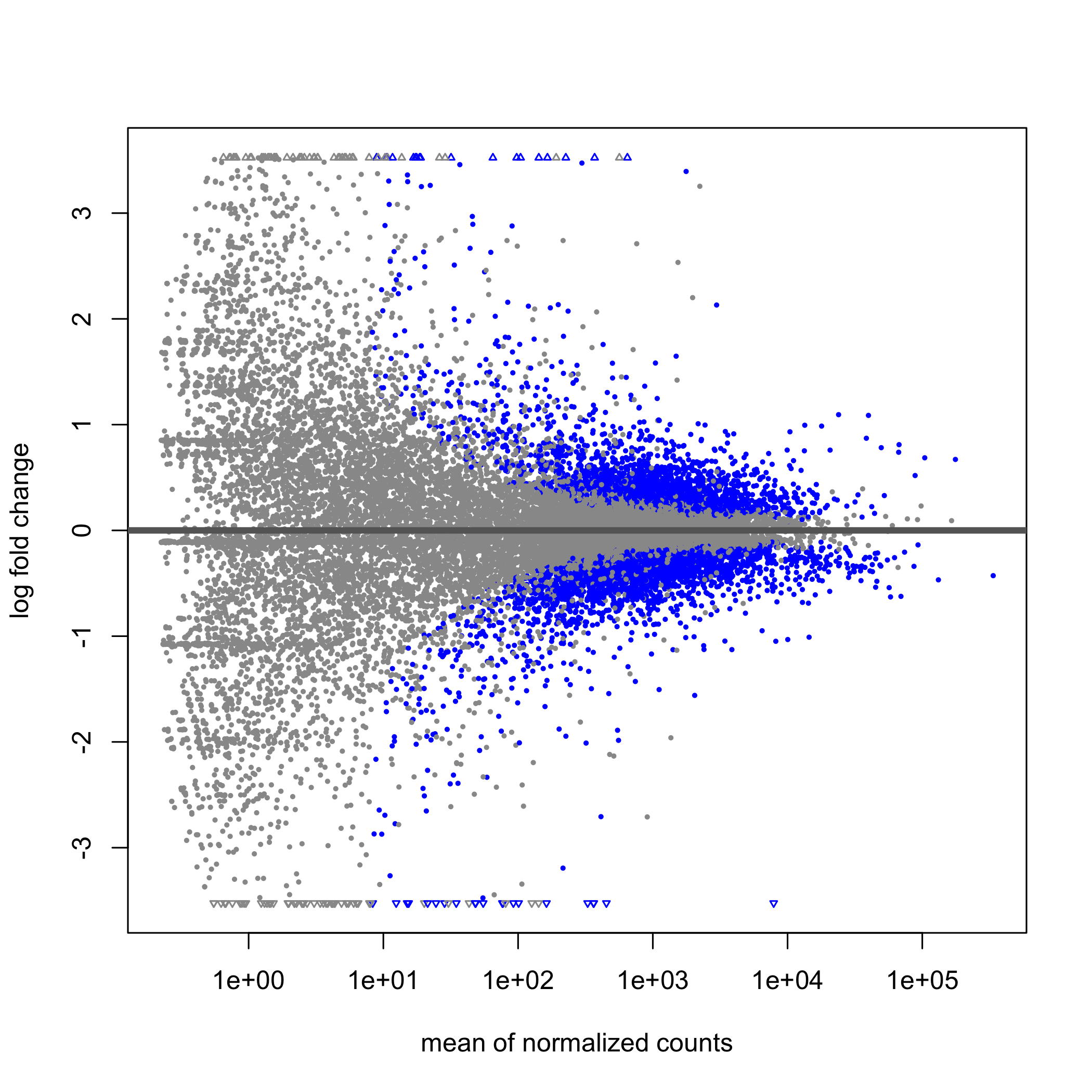<!-- --> ] ] --- # MAplot .scroll-output[ Para disminuir la sobre-estimación del LFC de genes que: - Son poco abundantes - Tienen alta dispersión Usemos la función `lfcShrink` de la librería *.red[apeglm]* ```r res_shrink <- lfcShrink(dds = dds, res = res, coef = "Treatment_siRNA_NRF2_vs_siRNA_control" , type = "apeglm") ``` ``` ## using 'apeglm' for LFC shrinkage. If used in published research, please cite: ## Zhu, A., Ibrahim, J.G., Love, M.I. (2018) Heavy-tailed prior distributions for ## sequence count data: removing the noise and preserving large differences. ## Bioinformatics. https://doi.org/10.1093/bioinformatics/bty895 ``` ```r summary(res_shrink) ``` ``` ## ## out of 21299 with nonzero total read count ## adjusted p-value < 0.1 ## LFC > 0 (up) : 2271, 11% ## LFC < 0 (down) : 2660, 12% ## outliers [1] : 26, 0.12% ## low counts [2] : 5778, 27% ## (mean count < 8) ## [1] see 'cooksCutoff' argument of ?results ## [2] see 'independentFiltering' argument of ?results ``` ```r plotMA(res_shrink, ylim = c(-3, 3)) ``` 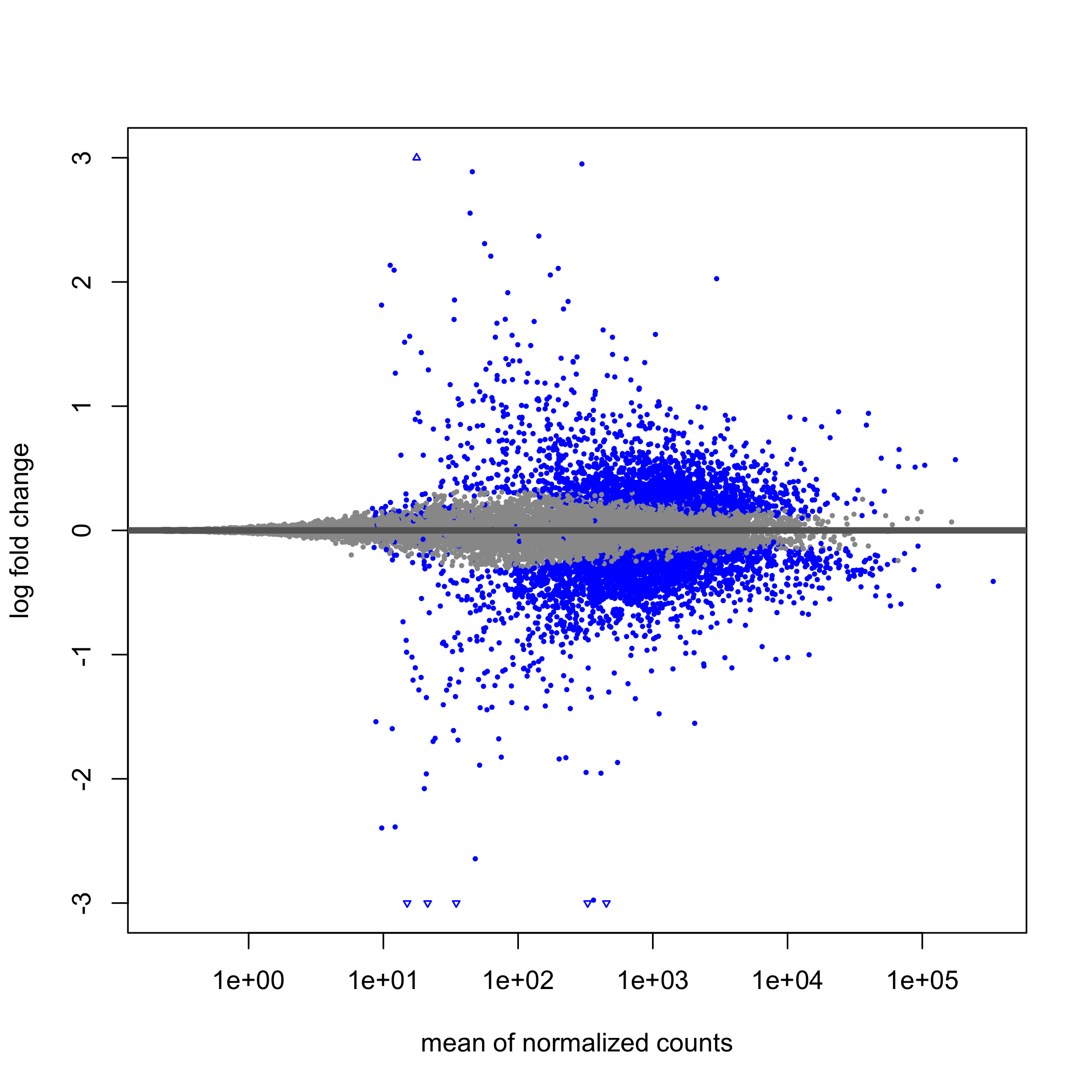<!-- --> ] --- # Volcano plot .scroll-output[ De manera similar al MAplot con el volcano plot visualizamos los genes que muestran expresión diferencial en nuestra condición de interés - En el eje y se grafica el -log10 de padj - En el eje x se grafica el lfc o *log2foldchange* Usa la función personalizada volcanoplotR del script de funciones: ```r ##En los argumentos logfc y padj utiliza los mismos valores empleados en la función results res_shrink <- as.data.frame(res_shrink) volcanoplotR(res_shrink, logfc = 0, p.adj = 0.1) ``` 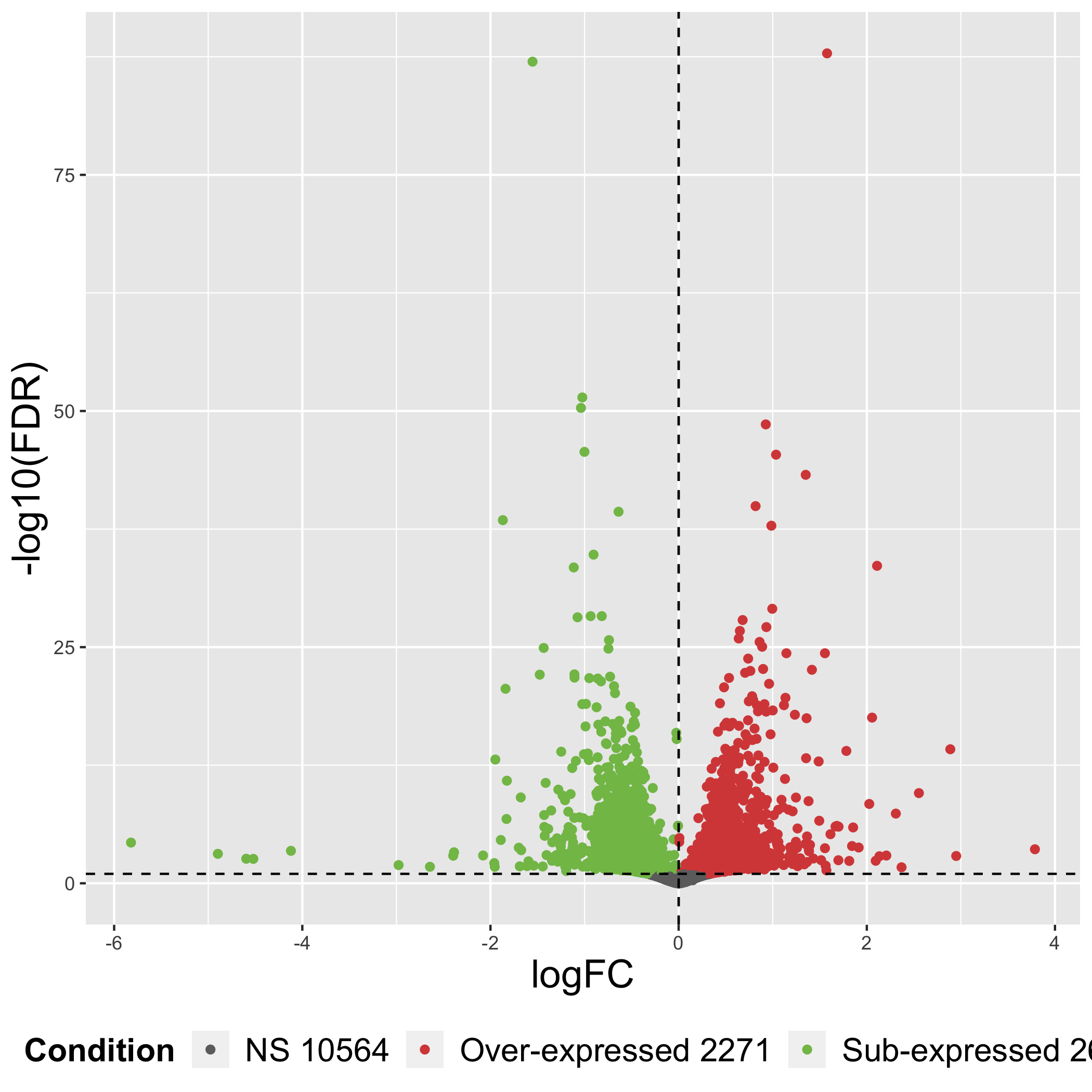<!-- --> ] --- ## Heatmap El *h eatmap* nos permite visualizar la expresión de los genes diferencialmente expresados en terminos de las cuentas normalizadas Consideraciones: - Usar los valores de las cuentas normalizadas para una mejor comparación entre muestras - Escalar los valores de las cuentas (renglones) para visualizar las diferencias en la expresión Usaremos la librería de *.red[pheatmap]* --- # Heatmap .scroll-output[ 1. Obten la matriz de cuentas normalizadas: ```r norm_counts <- counts(dds, normalized = T) ``` 2. Guarda un data.frame con los resultados de los genes mostrando expresión diferencial significativa: ```r deg <- res_shrink %>% dplyr::filter(log2FoldChange > 0 & padj < 0.1 | log2FoldChange < 0 & padj < 0.1) head(deg) ``` ``` ## baseMean log2FoldChange lfcSE pvalue padj ## ENSG00000000460 467.6023 0.4721205 0.11836789 9.277210e-06 0.0001217192 ## ENSG00000001084 1889.3028 -0.2991140 0.09464064 4.368589e-04 0.0031869719 ## ENSG00000001167 1068.3113 0.2413441 0.08978195 2.877301e-03 0.0149359373 ## ENSG00000001461 564.0667 -0.2490246 0.08409560 1.186278e-03 0.0073086983 ## ENSG00000001561 612.2146 -0.2536187 0.10372375 5.197697e-03 0.0240905329 ## ENSG00000001617 996.1836 0.2917830 0.09054617 3.973259e-04 0.0029527889 ``` 3. Usando la librería de `pheatmap()` obten la gráfica ```r ##Genera una paleta de colores RedBu <- brewer.pal(n = 10, name = "RdBu") RedBu <- rev(RedBu) pheatmap(norm_counts[rownames(deg), 1:6], ##Corta los genes con expresión diferencial de la matriz normalizada scale = "row", ##Calcula los valores z por fila (gen) color = RedBu, ##Nombre de la paleta de colores show_rownames = F, ##No mostrar el nombre de los genes en la gráfica show_colnames = F, ##No mostrar el nombre de las muestras en la gráfica clustering_distance_rows = "euclidean", ##Especifíca la medida de la distancia entre filas clustering_distance_cols = "euclidean", ##O columnas clustering_method = "single", ##Método de agrupamiento jerárquico border_color = NA, annotation_col = coldata[1:6, c(2, 6)]) ##Añadir información relacionada a las columnas) ``` ] ---